Publications
Click HERE for Sara's PubMed for the most up-to-date listing of publications.
Recent studies
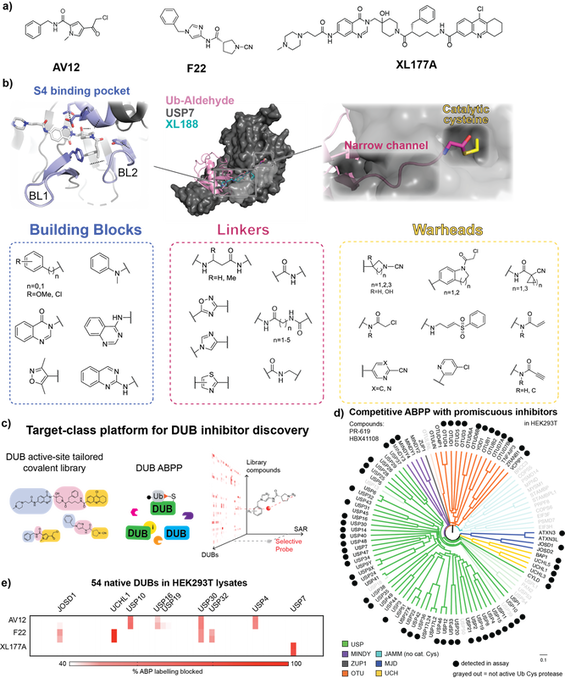 Chan, WC., Liu, X. et al. Nat Comms. 2023, 14: 686
Chan, WC., Liu, X. et al. Nat Comms. 2023, 14: 686
Accelerating Inhibitor Discovery for Deubiquitinating Enzymes
ABSTRACT: Deubiquitinating enzymes (DUBs) are an emerging drug target class of ~100 proteases that cleave ubiquitin from protein substrates to regulate many cellular processes. A lack of selective chemical probes impedes pharmacologic interrogation of this important gene family. DUBs engage their cognate ligands through a myriad of interactions. We embrace this structural complexity to tailor a chemical diversification strategy for a DUB-focused covalent library. Pairing our library with activity-based protein profiling as a high-density primary screen, we identify selective hits against 23 endogenous DUBs spanning four subfamilies. Optimization of an azetidine hit yields a probe for the understudied DUB VCPIP1 with nanomolar potency and in-family selectivity. Our success in identifying good chemical starting points as well as structure-activity relationships across the gene family from a modest but purpose-build library challenges current paradigms that emphasize ultrahigh throughput in vitro or virtual screens against an ever-increasing scope of chemical space.
Click HERE for the full paper
ABSTRACT: Deubiquitinating enzymes (DUBs) are an emerging drug target class of ~100 proteases that cleave ubiquitin from protein substrates to regulate many cellular processes. A lack of selective chemical probes impedes pharmacologic interrogation of this important gene family. DUBs engage their cognate ligands through a myriad of interactions. We embrace this structural complexity to tailor a chemical diversification strategy for a DUB-focused covalent library. Pairing our library with activity-based protein profiling as a high-density primary screen, we identify selective hits against 23 endogenous DUBs spanning four subfamilies. Optimization of an azetidine hit yields a probe for the understudied DUB VCPIP1 with nanomolar potency and in-family selectivity. Our success in identifying good chemical starting points as well as structure-activity relationships across the gene family from a modest but purpose-build library challenges current paradigms that emphasize ultrahigh throughput in vitro or virtual screens against an ever-increasing scope of chemical space.
Click HERE for the full paper
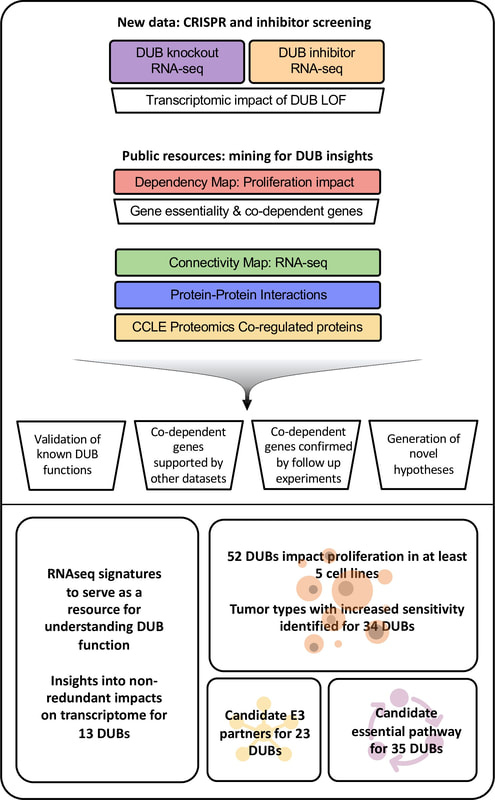 Doherty, LM et al. eLIfe. 2022, 11:e72879
Doherty, LM et al. eLIfe. 2022, 11:e72879
Integrating multi-omics data reveals function and therapeutic potential of deubiquitinating enzymes
ABSTRACT: Deubiquitinating enzymes (DUBs), ~100 of which are found in human cells, are proteases that remove ubiquitin conjugates from proteins, thereby regulating protein turnover. They are involved in a wide range of cellular activities and are emerging therapeutic targets for cancer and other diseases. Drugs targeting USP1 and USP30 are in clinical development for cancer and kidney disease respectively. However, the majority of substrates and pathways regulated by DUBs remain unknown, impeding efforts to prioritize specific enzymes for research and drug development. To assemble a knowledgebase of DUB activities, co-dependent genes, and substrates, we combined targeted experiments using CRISPR libraries and inhibitors with systematic mining of functional genomic databases. Analysis of the Dependency Map, Connectivity Map, Cancer Cell Line Encyclopedia, and multiple protein-protein interaction databases yielded specific hypotheses about DUB function, a subset of which were confirmed in follow-on experiments. The data in this paper are browsable online in a newly developed DUB Portal and promise to improve understanding of DUBs as a family as well as the activities of incompletely characterized DUBs (e.g. USPL1 and USP32) and those already targeted with investigational cancer therapeutics (e.g. USP14, UCHL5, and USP7).
Click HERE for the full paper
ABSTRACT: Deubiquitinating enzymes (DUBs), ~100 of which are found in human cells, are proteases that remove ubiquitin conjugates from proteins, thereby regulating protein turnover. They are involved in a wide range of cellular activities and are emerging therapeutic targets for cancer and other diseases. Drugs targeting USP1 and USP30 are in clinical development for cancer and kidney disease respectively. However, the majority of substrates and pathways regulated by DUBs remain unknown, impeding efforts to prioritize specific enzymes for research and drug development. To assemble a knowledgebase of DUB activities, co-dependent genes, and substrates, we combined targeted experiments using CRISPR libraries and inhibitors with systematic mining of functional genomic databases. Analysis of the Dependency Map, Connectivity Map, Cancer Cell Line Encyclopedia, and multiple protein-protein interaction databases yielded specific hypotheses about DUB function, a subset of which were confirmed in follow-on experiments. The data in this paper are browsable online in a newly developed DUB Portal and promise to improve understanding of DUBs as a family as well as the activities of incompletely characterized DUBs (e.g. USPL1 and USP32) and those already targeted with investigational cancer therapeutics (e.g. USP14, UCHL5, and USP7).
Click HERE for the full paper
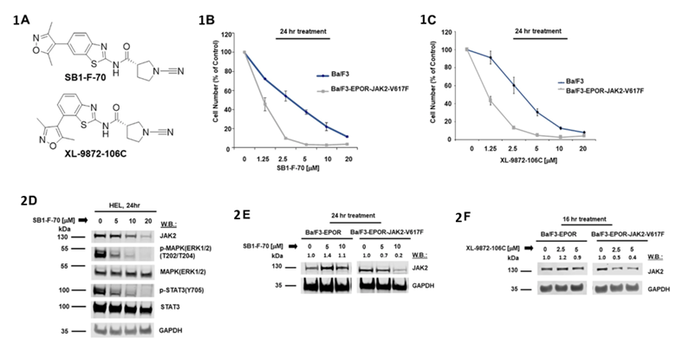 Yang, J. et al. Leukemia. 2022, 36, 210-220
Yang, J. et al. Leukemia. 2022, 36, 210-220
Small molecule inhibition of deubiquitinating enzyme JOSD1 as a novel targeted therapy for leukemias with mutant JAK2
ABSTRACT: Mutations in the Janus Kinase 2 (JAK2) gene resulting in constitutive kinase activation represent the most common genetic event in myeloproliferative neoplasms (MPN), a group of diseases involving overproduction of one or more kinds of blood cells, including red cells, white cells, and platelets. JAK2 kinase inhibitors, such as ruxolitinib, provide clinical benefit, but inhibition of wild-type (wt) JAK2 limits their clinical utility due to toxicity to normal cells, and small molecule inhibition of mutated JAK2 kinase activity can lead to drug resistance. Here, we present a strategy to target mutated JAK2 for degradation, using the cell's intracellular degradation machinery, while sparing non-mutated JAK2. We employed a chemical genetics screen, followed by extensive selectivity profiling and genetic studies, to identify the deubiquitinase (DUB), JOSD1, as a novel regulator of mutant JAK2. JOSD1 interacts with and stabilizes JAK2-V617F, and inactivation of the DUB leads to JAK2-V617F protein degradation by increasing its ubiquitination levels, thereby shortening its protein half-life. Moreover, targeting of JOSD1 leads to the death of JAK2-V617F-positive primary acute myeloid leukemia (AML) cells. These studies provide a novel therapeutic approach to achieving selective targeting of mutated JAK2 signaling in MPN.
Click HERE for the full paper
ABSTRACT: Mutations in the Janus Kinase 2 (JAK2) gene resulting in constitutive kinase activation represent the most common genetic event in myeloproliferative neoplasms (MPN), a group of diseases involving overproduction of one or more kinds of blood cells, including red cells, white cells, and platelets. JAK2 kinase inhibitors, such as ruxolitinib, provide clinical benefit, but inhibition of wild-type (wt) JAK2 limits their clinical utility due to toxicity to normal cells, and small molecule inhibition of mutated JAK2 kinase activity can lead to drug resistance. Here, we present a strategy to target mutated JAK2 for degradation, using the cell's intracellular degradation machinery, while sparing non-mutated JAK2. We employed a chemical genetics screen, followed by extensive selectivity profiling and genetic studies, to identify the deubiquitinase (DUB), JOSD1, as a novel regulator of mutant JAK2. JOSD1 interacts with and stabilizes JAK2-V617F, and inactivation of the DUB leads to JAK2-V617F protein degradation by increasing its ubiquitination levels, thereby shortening its protein half-life. Moreover, targeting of JOSD1 leads to the death of JAK2-V617F-positive primary acute myeloid leukemia (AML) cells. These studies provide a novel therapeutic approach to achieving selective targeting of mutated JAK2 signaling in MPN.
Click HERE for the full paper
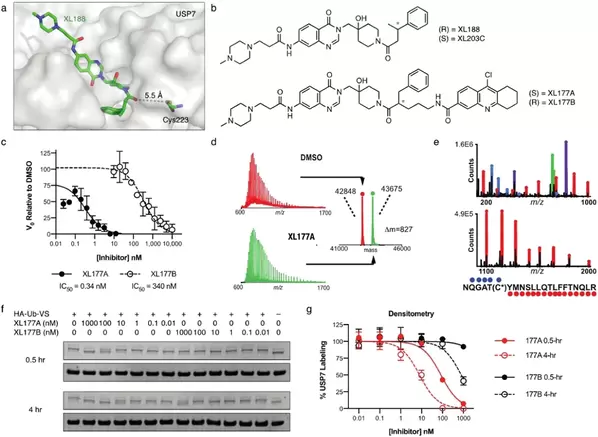 Schauer, N. et al. Scientific Reports 2020. 10, Article number: 5324
Schauer, N. et al. Scientific Reports 2020. 10, Article number: 5324
Selective USP7 inhibition elicits cancer cell killing through a p53-dependent mechanism
ABSTRACT: Ubiquitin specific peptidase 7 (USP7) is a deubiquitinating enzyme (DUB) that removes ubiquitin tags from specific protein substrates in order to alter their degradation rate and sub-cellular localization. USP7 has been proposed as a therapeutic target in several cancers because it has many reported substrates with a role in cancer progression, including FOXO4, MDM2, N-Myc, and PTEN. The multi-substrate nature of USP7, combined with the modest potency and selectivity of early generation USP7 inhibitors, has presented a challenge in defining predictors of response to USP7 and potential patient populations that would benefit most from USP7-targeted drugs. Here, we describe the structure-guided development of XL177A, which irreversibly inhibits USP7 with sub-nM potency and selectivity across the human proteome. Evaluation of the cellular effects of XL177A reveals that selective USP7 inhibition suppresses cancer cell growth predominantly through a p53-dependent mechanism: XL177A specifically upregulates p53 transcriptional targets transcriptome-wide, hotspot mutations in TP53 but not any other genes predict response to XL177A across a panel of ~500 cancer cell lines, and TP53 knockout rescues XL177A-mediated growth suppression of TP53 wild-type (WT) cells. Together, these findings suggest TP53 mutational status as a biomarker for response to USP7 inhibition. We find that Ewing sarcoma and malignant rhabdoid tumor (MRT), two pediatric cancers that are sensitive to other p53-dependent cytotoxic drugs, also display increased sensitivity to XL177A.
Click HERE for the full paper
ABSTRACT: Ubiquitin specific peptidase 7 (USP7) is a deubiquitinating enzyme (DUB) that removes ubiquitin tags from specific protein substrates in order to alter their degradation rate and sub-cellular localization. USP7 has been proposed as a therapeutic target in several cancers because it has many reported substrates with a role in cancer progression, including FOXO4, MDM2, N-Myc, and PTEN. The multi-substrate nature of USP7, combined with the modest potency and selectivity of early generation USP7 inhibitors, has presented a challenge in defining predictors of response to USP7 and potential patient populations that would benefit most from USP7-targeted drugs. Here, we describe the structure-guided development of XL177A, which irreversibly inhibits USP7 with sub-nM potency and selectivity across the human proteome. Evaluation of the cellular effects of XL177A reveals that selective USP7 inhibition suppresses cancer cell growth predominantly through a p53-dependent mechanism: XL177A specifically upregulates p53 transcriptional targets transcriptome-wide, hotspot mutations in TP53 but not any other genes predict response to XL177A across a panel of ~500 cancer cell lines, and TP53 knockout rescues XL177A-mediated growth suppression of TP53 wild-type (WT) cells. Together, these findings suggest TP53 mutational status as a biomarker for response to USP7 inhibition. We find that Ewing sarcoma and malignant rhabdoid tumor (MRT), two pediatric cancers that are sensitive to other p53-dependent cytotoxic drugs, also display increased sensitivity to XL177A.
Click HERE for the full paper